 A non-disruptive, highly automated, easily customizable approach to reducing storage costs and curating high quality data for AI.
A non-disruptive, highly automated, easily customizable approach to reducing storage costs and curating high quality data for AI.
Healthcare and life sciences organizations are sitting on billions of genomic sequencing files that hold insights to develop more effective treatments, precision medicine, and population health breakthroughs.
- BAM (Binary Alignment Map) and FASTQ files are stored across HPC clusters, on-premises NAS systems, object storage, and cloud environments. Analyzing them can compare gene expression, survey biodiversity, analyze DNA methylation, or investigate DNA-protein interaction, among many other next-generation sequencing applications.
- FASTQ files store DNA/RNA sequences and quality scores from sequencing machines. Analyzing these files can uncover accurate mutation detection, gene expression analysis, pathogen identification, and more.
Genomics pipelines generate massive data sets after sequencing workflows are complete, which while valuable also increase storage costs and complicate efficient search and curation for AI and BI tools.
Komprise AI Preparation and Process Automation (KAPPA) data services reduce the enterprise genomics storage footprint by identifying files that can be archived or deleted. KAPPA also delivers a rapid process to filter out low-quality data from research pipelines.
The hidden data problem in genomics sequencing data
BAM and FASTQ files have been a standard format for storing aligned sequencing reads for over a decade. They were designed for storage and bioinformatics tool compatibility, not for analytics or AI at scale.
These files carry detailed contextual metadata embedded in file headers, including average quality score, average read length, project ID, read count, sequencing platform, reference genome version, sample identifiers and alignment statistics. But that metadata is invisible to the storage layer. What the file system sees is a filename, a size, a modification date, and an owner: nothing about the patient, the sequencing run, the tissue type, or the variant calling workflow that produced it.
This is the core problem:
- Genomics pipelines generate large temporary files (often 10 to 100+ GB per compressed file) that are stored on expensive flash storage after sequencing workflows are complete;
- Data storage is one of the largest cost line items in genomics research;
- IT directors can’t delete or archive this data without insight into data usage and access;
- There is no efficient way to search, filter, or tag BAM and FASTQ files based on biological or clinical attributes, slowing research progress and degrading quality.
Meanwhile, demand for genomics AI continues to grow. Sequencing and variant analysis are among the top AI use cases in precision medicine and clinical research.
The global genomics market is projected to reach over $94 billion by 2030, driven largely by AI-powered analysis of sequencing data.
What genomic AI data preparation requires
Clinical and research AI depends on access to clean, high-quality data with precise biological context.
Preparing genomic data for AI typically involves:
- Culling old, stale, low authority and duplicate data from analytics pipelines;
- Extracting header metadata such as sample ID, reference genome, sequencing platform, library preparation method, and read group details;
- Tagging files so they can be cataloged, searched, and governed across research and clinical teams;
- Exporting structured metadata into formats such as Apache Iceberg tables for scalable analytics and cohort assembly;
- Curating datasets and making them accessible to AI pipelines, genome browsers, and BI tools.
This process must happen without disrupting research workflows, without requiring changes to existing bioinformatics infrastructure, and without creating persistent duplicate copies of data that inflate storage costs. This is a significant concern given that a single whole genome sequence can generate 100GB or more of raw data.
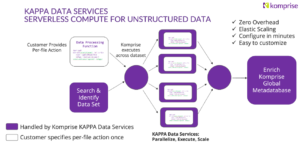
KAPPA Data Service for Genomics
KAPPA is a serverless compute framework for unstructured data that allows organizations to run custom functions directly on files in place. With just a few lines of Python code, teams can define how data should be processed, without provisioning infrastructure or building complex pipelines. Read more.
These are the top use cases for KAPPA in genomics:
- Data storage optimization: An IT infrastructure administrator can see all interim BAM and FASTQ files for specific completed projects with the enriched metadata from KAPPA data services. They can then filter this data set by time of last access, such as 30, 90 or 365 days after the project completion dates and tier the data from hot storage to a cold archive in the data center or in the cloud. This way, just the interim files for completed projects are aged out by policy. Similarly, Komprise can search for and extract metadata indicating duplicate runs and confine these data sets for deletion. This process can happen automatically and continuously.
- Genomics data classification: KAPPA can use a script to tag data with clinical labels such as sequencing platform, read count, and average read length and then move those curated data sets to AI and analytics tools. Once tagged, bio-informaticians can easily find data over and again as new studies arise.
- Filter data sets for quality: You can also use KAPPA to filter runs that are too short, too small, or from the wrong instrument before they are ingested by the AI platform. This is a prominent use case as sequencing labs produce hundreds of FASTQ files per run, a high percentage of which are irrelevant and needlessly clog processing and storage resources.
What KAPPA means for genomics AI and research
The result is that genomic data that was previously opaque becomes AI-ready. Research and clinical AI teams can now search and filter datasets using real biological criteria. For example, they can identify whole genome sequences from a specific patient cohort, RNA-sequence runs using a particular library kit, or aligned reads mapped to a specific reference genome version. And they can do this without moving a single file or spending months on custom data engineering.
Benefits of the serverless Komprise approach
KAPPA handles the processing and infrastructure at scale, without touching existing research applications and without relying on brittle scripts that are hard to maintain as file volumes grow. KAPPA operates as a layer above the storage environment, not inside the application stack. Importantly, the extracted metadata persists even if the underlying data moves across storage tiers, ensuring that AI pipelines can always find and trust the data they need.
Organizations gain:
- Cost-efficient data management, eliminating temporary and unneeded data from the high-performance storage tier;
- Faster genomic dataset discovery and cohort assembly;
- Improved research productivity and reduced time from sequencing to insight;
- Easier dataset curation for AI model training and validation;
- Direct access for AI agents, variant calling pipelines, and machine learning platforms.
Why this matters now
As genomic AI becomes more prevalent across oncology, rare disease research, and pharmacogenomics, IT infrastructure teams are under growing pressure to deliver AI-ready sequencing data on demand. That means supporting more requests from research and clinical teams, maintaining governance, controlling runaway storage costs of petabyte-scale sequencing archives, and avoiding disruption to existing bioinformatics workflows.
The takeaway
KAPPA data services provide a practical way to meet AI data workflow and preparation requirements for genomics by automating BAM and FASTQ metadata extraction and turning unstructured sequencing data into structured, searchable, and governed datasets.
It allows healthcare and life sciences organizations to prepare genomics data for AI without wasting expensive data storage resources and without changes to existing sequencing or analysis infrastructure. In doing so, it transforms genomic sequencing data from something that is simply stored into something that can be actively used for AI, research collaboration, and precision medicine at scale.
KAPPA data services are currently in an early access program for customers. Contact your Komprise account team to discuss specific use cases. To schedule a demonstration and learn more visit: Komprise.ai/KAPPA
Learn more about Komprise for Life Sciences & Genomics.


